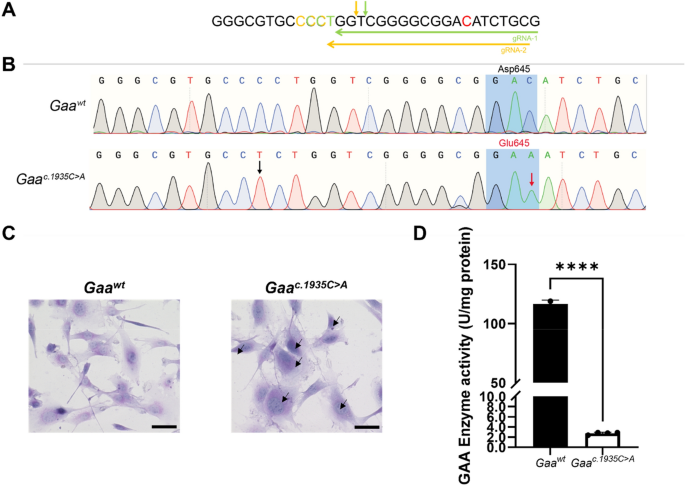
Pompe disease, an autosomal recessive disorder caused by deficient lysosomal acid α-glucosidase (GAA), is characterized by accumulation of intra-lysosomal glycogen in skeletal and oftentimes cardiac muscle. The c.1935C>A (p.Asp645Glu) variant, the most frequent GAA pathogenic mutation in people of Southern Han Chinese ancestry, causes infantile-onset Pompe disease (IOPD), presenting neonatally with severe hypertrophic cardiomyopathy, profound muscle hypotonia, respiratory failure, and infantile mortality. We applied CRISPR-Cas9 homology-directed repair (HDR) using a novel dual sgRNA approach flanking the target site to generate a Gaaem1935C>A knock-in mouse model and a myoblast cell line carrying the Gaa c.1935C>A mutation. Herein we describe the molecular, biochemical, histological, physiological, and behavioral characterization of 3-month-old homozygous Gaaem1935C>A mice. Homozygous Gaaem1935C>A knock-in mice exhibited normal Gaa mRNA expression levels relative to wild-type mice, had near-abolished GAA enzymatic activity, markedly increased tissue glycogen storage, and concomitantly impaired autophagy. Three-month-old mice demonstrated skeletal muscle weakness and hypertrophic cardiomyopathy but no premature mortality. The Gaaem1935C>A knock-in mouse model recapitulates multiple salient aspects of human IOPD caused by the GAA c.1935C>A pathogenic variant. It is an ideal model to assess innovative therapies to treat IOPD, including personalized therapeutic strategies that correct pathogenic variants, restore GAA activity and produce functional phenotypes. - READ MORE
By Nature.com
Wed, 14 Dec 2022 10:04:03 GMT


:quality(70)/cloudfront-us-east-1.images.arcpublishing.com/archetype/X3HXZODDPFHXVMJEKMJ4KG2RS4.jpg)